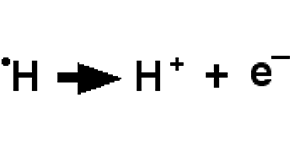Université de Liège - Centre de l'Oxygène, Recherche et Développement (CORD)
Université de Liège - Centre de l'Oxygène, Recherche et Développement (CORD)
L'oxygène et la vie: tome 1 - Intitiation au métabolisme de l'oxygène
C Deby et G Deby-Dupont
Université de Liège - Centre de l'Oxygène, Recherche et Développement (CORD)
L'oxygène et la vie: tome 1 - Intitiation au métabolisme de l'oxygène
C Deby et G Deby-Dupont
Chapitre III: Introduction physico-chimique très élémentaire
1. Atomes et molécules
Les atomes et les molécules sont des édifices compliqués dont la réunion et l'agencement constituent la matière. Dans les atomes, on distingue des électrons, particules chargées d'électricité négative, dispersées par la répulsion qu'elles exercent entre elles (car étant de même signe), mais maintenues dans un espace bien délimité par l'attraction d'un noyau dont la charge électrique positive est :
- soit égale à la somme des charges des électrons (atome neutre),
- soit supérieure d'une ou de plusieurs unités (ions positifs ou cations) ou inférieure d'une ou de plusieurs unités (ions négatifs ou anions) à cette somme.
L'énergie cinétique des molécules et des atomes constituant celles-ci augmente avec la température. Nulle au zéro absolu (- 273°C), elle s'élève avec la température: la molécule commence à vibrer. A partir d'une certaine température, à cette vibration s'ajoute une rotation.
La distribution des électrons autour du noyau est faite selon divers niveaux énergétiques, dans des "cases" énergétiques que l'on nomme orbitales et qui ont remplacé la vieille image d'orbite planétaire qui fut proposée il y a une centaine d'années, mais qui a été définitivement abandonnée au cours de la seconde moitié du XXe siècle. Dans ces "cases", il y a place pour 2 électrons au maximum (Principe de Pauli). Les électrons sont le plus souvent répartis par paire (structure stable).
Représentation de Lewis pour les atomes légers : il s'agit d'un modèle un peu simplifié, mais aisé pour comprendre la plupart des interactions en chimie biologique. Lewis ne représentait que les électrons "extérieurs", ceux qui sont impliqués dans les réactions chimiques (électrons de valence) et qui ne peuvent être au maximum que 8. C' est la règle de l'octet de Lewis, applicable aux atomes légers, principaux constituants de la matière vivante, comme par exemple le carbone, l'azote, l'oxygène, le soufre, etc...
Dans les réactions, les atomes gagnent ou perdent des électrons pour atteindre ce nombre magique, 8, gage de stabilité des édifices moléculaires d'atomes de petite taille.

2. Para- et diamagnétisme
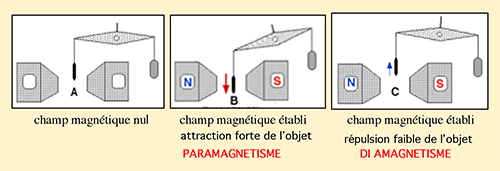
| Fig. III-1 : Para - et diamagnétisme A : Un objet est suspendu à une balance à proximité de l'entrefer d'un électro-aimant non branché à une source de courant. B et C : Si on établit un champ magnétique en connectant l'électro-aimant à une source de courant continu, l'objet, selon sa nature, peut avoir deux comportements : • cas B : être attiré au centre du champ ; il peut alors être classé parmi les corps paramagnétiques (métaux ferreux par exemple) • cas C : il est repoussé par le champ magnétique ; il est constitué par des substances diamagnétiques (métaux non ferreux ; la plupart des substances organiques, etc). |
L'attraction exercée sur un corps paramagnétique est beaucoup plus forte que la répulsion agissant sur une substance diamagnétique : le diamagnétisme passe ainsi généralement inaperçu et nécessite un appareillage sensible pour être constaté.
3. Le magnétisme de l'électron
NB : les comparaisons entre les concepts quantiques et les objets à notre dimension ont été assumées par les physiciens à l'aube du XXe siècle et de la "nouvelle" physique et constituent une approximation très utile dans un exposé didactique élémentaire.
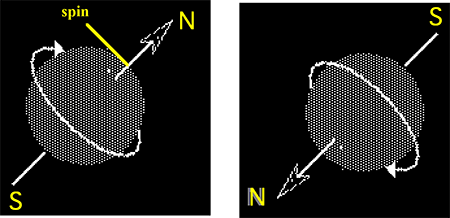
Fig. III-2: Les deux états de l'électron : Vers 1920, on assimilait les électrons à des toupies chargées électriquement qui, tournant sur elles-mêmes, engendraient un champ magnétique orienté perpendiculairement au plan de rotation (flèche blanche); le vecteur champ (spin) est orienté selon la règle des trois doigts. Comme l'électron ne peut tourner que dans deux sens opposés, il n'y a que deux orientations possibles pour le spin. Electron : intuitivement, ce quantum porteur d'une charge électrique négative peut être comparé à une toupie tournant sur elle-même et qui, de par sa charge, engendre un vecteur magnétique perpendiculaire au sens de la rotation. L'électron peut donc aussi être assimilé à une aiguille aimantée influençable par un champ magnétique (expérience d'Oersted : 1830). Son orientation dans un champ magnétique externe est parallèle à l'axe Nord-Sud de ce dernier. Son sens dépend du sens de la rotation de l'électron : il n'y a que deux possibilités puisqu'il n'y a que deux sens de rotation possibles. Noter qu'à ce vecteur magnétique s'ajoute le moment cinétique, vecteur mécanique parallèle ou antiparallèle au premier.
Il faut fournir de l'énergie pour changer la direction du spin (similitude avec le gyroscope).Valeurs du spin : calculé en fonction de la constante quantique universelle hν, il y a deux valeurs pour le spin électronique :
s = 1/2 h/2π et s = -1/2 h/2π, ou plus simplement 1/2 et -1/2, de même qu'il n'y a que deux sens possibles de rotation autour d'un axe pour une toupie.
L’existence d’orbitales à un seul électron est caractéristique des radicaux libres: la réactivité de ceux-ci est due à l’attraction d'un autre électron lié à une autre molécule, dont le sens de rotation est inverse de celui de l'électron occupant l'orbitale en question (ce qui entraîne un spin inverse).
Dans une orbitale remplie, les spins ont leurs vecteurs opposés et la somme des spins est (+1/2) + (-1/2) = 0. Une molécule à l'état fondamental est généralement non paramagnétique (sauf l'oxygène).
4. Liaison par covalence

Dans le cas de l'oxygène, les électrons périphériques sont au nombre de 6 pour l'atome et de 12 pour la molécule O2; la couche de Lewis de cet élément est donc incomplète.
Dans la liaison par covalence, deux électrons sont appariés, leurs spins étant opposés (fig. III-3), ce qui annule leurs champs magnétiques respectifs.
Dans une molécule dont tous les électrons sont appariés, la résultante magnétique est nulle (principe de Pauli ; voir plus bas : Multiplicité).
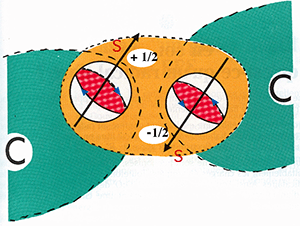
Fig. III-3: Représentation extrêmement simplifiée du mécanisme de la covalence unissant deux atomes de carbone C.
Les électrons tournent sur eux-mêmes: comme ils sont électriquement chargés, cette rotation engendre un champ magnétique, perpendiculaire au plan de rotation, symbolisé par un vecteur S, le spin. Deux orientations opposées sont possibles, dépendant du sens de la rotation de l'électron et sont désignées arbitrairement par + S et -S. La liaison par covalence est réalisée par la mise en commun, par deux atomes, de deux électrons de spins opposés (donc de sens inverse de rotation) qui seront localisés sur une même orbitale.
5. Les structures radicalaires
a. Considérations générales
La chimie organique, dès ses débuts, a désigné sous l'appellation de radical la partie non active de la molécule qui supporte une fonction ; par exemple : CH3-OH, CH3CH2-OH, etc ; dans ces cas-ci, on voit que la fonction OH (alcool) est supportée par des chaînes de plus en plus longues, les radicaux, auxquels, par souci de classification, on donne le nom de l'hydrocarbure correspondant, avec la terminaison -yle. Ainsi, CH3- dérive du méthane CH4 et est nommé radical méthyle d'où dériveront l'alcool méthylique, le chlorure de méthyle, si OH est remplacé par Cl, etc ... La notion de radical, à l'aurore de la chimie moderne, faisait simplement partie d'un système de classification qui facilitait l'enseignement de la chimie organique et permettait d'établir une meilleure nomenclature.
Mais des pionniers comme Gombert ont soutenu que ces entités purement didactiques devaient exister sans être liées à une fonction : CH3, CH3CH2, etc... Le concept de radical libre était né. Des décennies durant, la théorie des radicaux libres fut combattue. Mais simultanément se développait la chimie quantique. Elle est apparue vers la moitié du XXe siècle dans la chimie avancée et a triomphé définitivement depuis la décennie 60.
Mais une aura mystérieuse envelopperait pendant longtemps ces évanescents radicaux libres auxquels les biologistes et les médecins, sensibles à la science-fiction, accorderaient des propriétés diaboliques: "on n'adore que l'incompris que l'on nomme absolu"...
Aujourd'hui, l'existence des radicaux libres n'est plus mise en doute par les chimistes. Il s'agit d'édifices atomiques dont la couche d'électrons de valence est impaire, par existence d'une orbitale non remplie. Ils sont donc porteurs d'un électron non apparié dont le spin n'est pas annulé par un électron de signe contraire. Ils sont donc obligatoirement paramagnétiques.
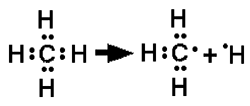
Fig. III-4 : Création d'un radical libre (méthyl) à partir du méthane, par enlèvement d'un atome d'hydrogène (rupture d'une covalence C-H ): présence d'un électron célibataire sur l'atome de carbone. CH3• est un radical extrêmement réactionnel. H• est également un radical mais qui cède immédiatement son électron à une molécule réductible pour devenir le banal proton H+. Cette réaction nécessite de l'énergie qui est fournie dans le vivant par des enzymes. Ces molécules sont généralement dotées d'une grande réactivité, car elles ont tendance à remplir leur orbitale déficitaire en captant ailleurs un électron, souvent porté par un atome tel qu'un hydrogène qui est arraché à une molécule, laquelle devient à son tour radicalaire.
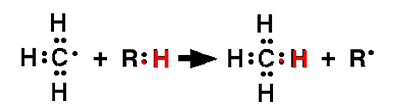
| Fig. III-5 : Cette réaction est spontanée si le nouveau composé formé est plus stable que RH. |
La réaction de la fig. III-5 a produit un radical R• qui pourra réagir avec une nouvelle molécule Y:H, si R:H est plus stable que Y:H, etc ... La chaîne de réactions radicalaires s'arrêtera si deux radicaux dimérisent :
R• + R• ===> R:R
ou si le dernier radical formé rencontre une molécule donnant naissance à un radical très stable. C'est le cas de la vitamine C (voir au chapitre IV, la fig. IV-9 et dans ce chapitre, les réactions en chaîne, fig. III-10).
Importance de la durée de vie d'un radical : la résonance
On admet que, pour jouer un rôle modificateur dans la biochimie d'une cellule, le radical libre doit pouvoir exister suffisamment de temps pour quitter le lieu de sa genèse et atteindre des cibles plus ou moins éloignées (nous parlons évidemment d'un maximum de quelques dizaines d'angströms).
On admet que le radical •OH a une durée de vie inférieure à la nanoseconde et qu'il se combine immédiatement avec une molécule voisine ou se recombine selon la réaction :
HO• + •OH ==> HO:OH ou H2O2
pour former l'eau oxygénée.
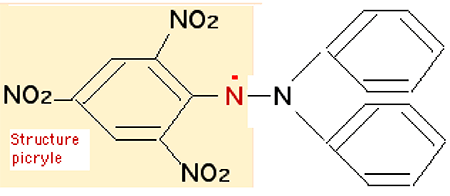
En dehors des phénomènes de radiolyse (voir ci-dessous), il n'y a aucune certitude que •OH existe réellement, même pendant une nanoseconde. Mais la structure de la molécule porteuse d'un électron célibataire peut allonger considérablement la durée de vie de l'état radicalaire. On peut isoler et purifier des molécules radicalaires comme le diphénylpicrylhydrazyl (fig. III-5 bis).
Fig. III-5 bis : Une molécule radicalaire: le diphénylpicrylhydrazyl.Ce composé, pourtant radicalaire, est très stable à l'état cristallisé et "vit" des dizaines de minutes en solution dans l'alcool. L'explication de cette stabilité est fournie par le phénomène de délocalisation de l'électron célibataire. Cet électron, originaire de N, voyage sur toute la structure picryle : il est en état de liberté très contrôlée qui retarde ses chances de rencontre avec l'électron célibataire d'un éventuel radical libre (ou un biradical) étranger (par exemple, l'oxygène).
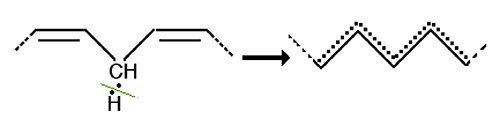
Fig. III-6 : Constitution d'une structure résonante.
La fig. III-6 montre comment se constitue une structure à électron délocalisé ou résonante. Dans une chaîne carbonée, des liaisons simples alternent avec des doubles liaisons. Si on enlève un hydrogène muni de son électron en un point de cette chaîne, le carbone concerné portera un électron non apparié qui va se délocaliser sur une distance de plusieurs carbones, réduisant la probabilité de sa rencontre avec un atome d'oxygène en fonction de ce nombre d'atomes de C. On voit que la "semi-liberté" de l'électron célibataire réduit sa cinétique de réaction et sa "dangerosité" ! A la délocalisation correspond une diminution de la réactivité qui devient plus sélective. Une telle molécule radicalaire peut se déplacer sur une distance de quelques microns.
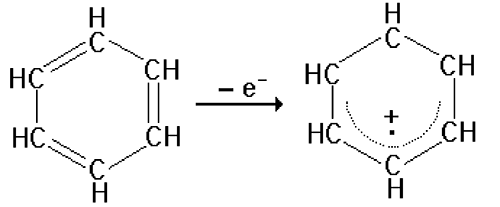
Fig. III-7 : Autre exemple de structure radicalaire résonante : le cation benzoyle, formé en enlevant un électron à la molécule de benzène : l'électron célibataire est délocalisé sur tout le cycle. b. Intérêt des radicaux libres en biologie
En 1895, Röntgen découvrait les rayons X, produits par des faisceaux d'électrons créés en atmosphère raréfiée, dans des champs électriques continus de plusieurs dizaines de milliers de volts. Ces rayons ont la propriété de traverser des matières opaques à la lumière visible. L'année suivante, Becquerel démontrait l'émission de radiations aussi pénétrantes que les rayons X, émises par les sels d'uranium : les rayons gammas (γ). Le même constatait une ionisation de l'air autour de ces sels. Cette propriété sera utilisée par Pierre et Marie Curie qui, munis d'électroscopes, isoleront en 1898 le polonium et le radium. Promptement, le caractère toxique de ces radiations sera soupçonné : en 1905, Pierre Curie dénonce le danger au cours d'une conférence Nobel. Le médecin radiologue Bergonié paiera l'un des premiers son tribut aux radiations ionisantes en devant subir des amputations successives. Mais les principes de radioprotection resteront inconnus durant l'entre-deux guerres ; il faudra les atroces expériences d'Hiroshima et de Nagasaki pour que l'on prenne conscience du mal des rayons. C'est alors que commence vraiment la radiobiologie. C'est le projet Manhattan (mise au point de bombes atomiques, commencée en 1940) qui permettra de découvrir deux phénomènes capitaux pour la compréhension des radiolésions :
a) l'eau irradiée, par rayons X ou γ, se décompose avec formation de radicaux libres •H et •OH (radiolyse de l'eau),
b) la radiolyse de l'eau augmente avec la pression d'oxygène (effet oxygène).
Déjà avant la guerre de 1914, le français Debierne avait reconnu que la décomposition de l'eau par les rayonnements dits ionisants implique la formation d'espèces instables.
Après 1945, radiochimie et radiobiologie se développeront parallèlement. Une explication du mécanisme des lésions occasionnées par les radiations ionisantes est fournie: l'énergie libérée dans les milieux absorbant les rayonnements X ou γ dissocie les covalences et crée des paires de radicaux.
Comme l'eau constitue la majeure partie de la matière vivante, elle subit la radiolyse et libère au sein des cellules, non seulement des radicaux hydroxyles •OH, dont les radiochimistes ont démontré l'extrême réactivité, mais d'autres espèces radicalaires et par recombinaison des radicaux •OH et de l'eau oxygénée H2O2 (voir plus bas l'importance de cette espèce oxygénée).
Nous expliquons l'effet oxygène dans le chapitre V.A partir des travaux de Fridovich (1968), les biochimistes envisagèrent que des radicaux •OH pouvaient être créés au sein de la matière vivante par des agents autres que les radiations ionisantes. Rapidement deux camps divisèrent les chercheurs : d'un côté les médecins, biologistes et biochimistes, de l'autre, les chimistes et physicochimistes, ces derniers affirmant que les mécanismes de production d'•OH invoqués par les adeptes des sciences de la vie (réactions de Fenton et d'Haber-Weiss) n'étaient pas thermodynamiquement possibles. La controverse dure toujours ! Si les physicochimistes admettent que des radicaux libres peuvent apparaître in vivo, ils invoquent d'autres mécanismes et d'autres espèces oxygénées activées, telles que les oxo-ferryles (voir chapitre VIII).
De toute manière, une nouvelle orientation était donnée à la biochimie, faisant intervenir de manière plus générale l'élément oxygène.Les radicaux libres et le mythe de l'électron libre
Dans l'imagerie d'une certaine presse de vulgarisation, l'électron solitaire dans une orbitale est présenté comme un "agent incontrôlé".
L'expression "électron libre" a été fâcheusement introduite dans le langage familier lors de la guerre des Balkans, en 1991. L'idée est maintenant répandue dans un certain public que de tels agents surgissent dans l'organisme et s'introduisent subrepticement dans les rouages des mécanismes vivants, à la manière des terroristes. Il est certain que les électrons libres n'existent pas dans les conditions biologiques de température ; ils sont toujours liés à un noyau atomique; ces particules ne deviennent libres que dans le quatrième état de la matière, le plasma, qui n'existe qu'aux très hautes températures ainsi que dans la couche conductrice de certains solides.
Solitaires ou appariés, les électrons restent étroitement assujettis à l'atome dont ils font partie et ne le quitteront que pour entrer immédiatement dans la sphère d'influence d'un autre atome (Principes de Frank-Condon, fig. III-20, et lois de Marcus).Le terme multiplicité trouve son origine dans la spectroscopie.
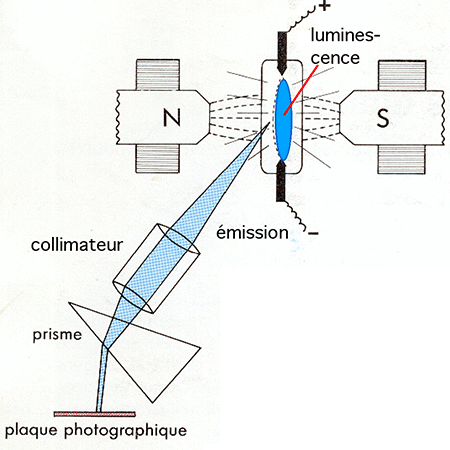
Les recherches de base de la mécanique quantique reposent en grande partie sur l'étude de la lumière émise par des gaz raréfiés soumis à un champ électrique de tension élevée (tubes à décharge, ampoules de Crookes, etc ...). Cette lumière témoigne de l'excitation des atomes gazeux provoquée par ce champ. Le spectre de la lumière peut être décomposé par un prisme ou un réseau et se résout en raies caractéristiques de l'élément ou du composé expérimenté. Certaines raies du spectre peuvent être modifiées par la superposition, au champ électrique, d'un champ magnétique. La figure III-8 montre un exemple de dispositif expérimental qu'on utilisa pour ce type de recherche pionnière. Les spectres étaient reçus sur des plaques photographiques (spectrographes).
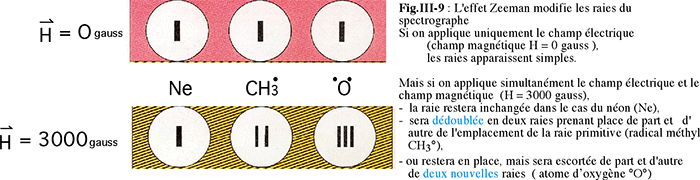
Fig. III-8 : Effet Zeeman : dispositif expérimental.La figure III-9 montre 3 raies choisies pour une molécule et deux éléments : le néon, le radical méthyle (formé à partir du méthane soumis à une haute tension) et l'oxygène monoatomique (provenant de la séparation des deux atomes O2 par la haute tension).
On dit que le néon est un singulet (singlet) car la raie envisagée est restée unique, que le radical méthyle est un doublet, tandis que l'oxygène, dont la raie caractéristique est devenue triple, est qualifié de triplet.Ces différents cas, révélés par l'effet Zeeman, définissent la multiplicité de l'atome ou de la molécule.
Plus tard, les physiciens ont démontré que les singulets ne présentent aucun électron célibataire, tandis que les doublets et les triplets en présentent respectivement 1 et 2 (il existe des états multiplets dont il ne sera pas question ici).


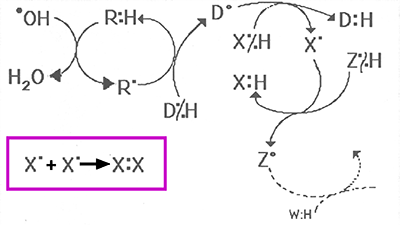
Fig. III-10 : Exemple de réactions en chaîne. En encadré : recombinaison de deux radicaux
libres qui interrompent la chaîne (réaction moins rapide : voir plus bas).
Le plus agressif des radicaux libres est le radical hydroxyle •OH, généré par la radiolyse de l'eau. Dans notre exemple, •OH arrache un hydrogène accompagné de son électron à la molécule organique RH, qui devient à son tour un radical libre R•, tandis que le radical hydroxyle devient de l'eau. R• peut arracher un H• à une autre molécule, telle DH, qui devient elle-même un radical libre D•, et ainsi de suite ...
Mais la réaction peut s'arrêter par dimérisation (réaction encadrée), deux radicaux couplant leurs spins pour former un dimère tel que X-X, ou D-D, etc ...
Mais ces réactions de dimérisation ne sont pas favorisées (règle de Wigner) et demandent plus de temps à se produire que les réactions radical + molécule "neutre" (singulet). Ces réactions en chaînes peuvent donc durer un temps relativement long. La présence d'oxygène vient compliquer et allonger les voies de réaction et leur durée (voir chapitre X, fig. X-4).e. La résonance paramagnétique électronique (RPE)(ultra-simplifiée)
1. Généralités : une structure chimique susceptible de renfermer un ou plusieurs électrons célibataires est introduite dans une enceinte réfléchissant des ondes électromagnétiques stationnaires, de fréquence très élevée (en d'autres termes, de longueurs d'onde centimétriques, ou microondes). La RPE utilise des microondes de fréquence fixe (généralement 9,2 gigahertz).
Cette enceinte est reliée par un guide d'onde à un détecteur susceptible d'enregistrer une diminution de l'énergie dans ce système stationnaire. L'enceinte porteuse de l'échantillon est installée dans l'entrefer d'un puissant électro-aimant dont le champ croît progressivement. Si l'échantillon renferme des structures radicalaires, on constate que, pour une valeur précise du champ magnétique, il y a absorption de l'énergie de la microonde, grâce à un détecteur qui enregistre un pic d'absorption qui passe par un maximum lorsque la valeur de résonance du champ magnétique est atteinte pour un radical donné. Ensuite, l'intensité du champ magnétique continuant à croître, l'absorption diminue et l'équilibre antérieur est retrouvé. La RPE permet non seulement de montrer l'existence de structures radicalaires dans un échantillon, mais, plus encore, d'identifier la molécule porteuse d'électron célibataire (fig. III-13).
Nous voyons sur la fig. III-11 que le spectre d'absorption est un pic (A) dont on préfère enregistrer la dérivée première B (explication du caractère biphasique des tracés), ce qui facilite, par son augmentation d'amplitude, la mesure de l'absorption.
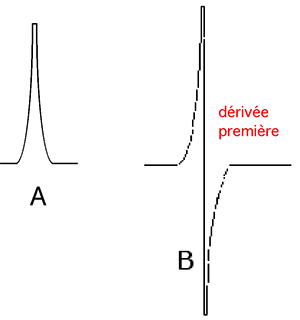
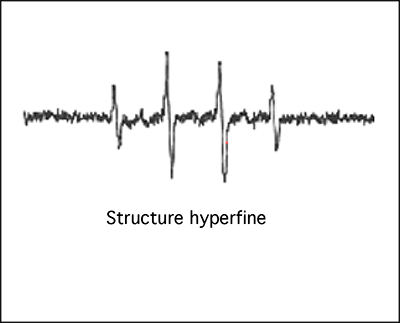
Fig. III-11 Fig. III-12
2. Structure hyperfine : la simplicité de la fig. III-11 est loin d'être la règle pour les radicaux organiques. Il existe en effet des interactions entre les spins nucléaires et électroniques, engendrant des clivages du pic initial : c'est la structure hyperfine du spectre EPR (fig. III-12) où de nombreuses raies apparaissent.
L'interprétation de cette structure compliquée se fait notamment par l'utilisation de "constantes de couplage hyperfin".
Un exemple très commun de structure hyperfine est fourni par des échantillons de sang où l'on peut observer le spectre du radical ascorbyle (fig. III-13 A).
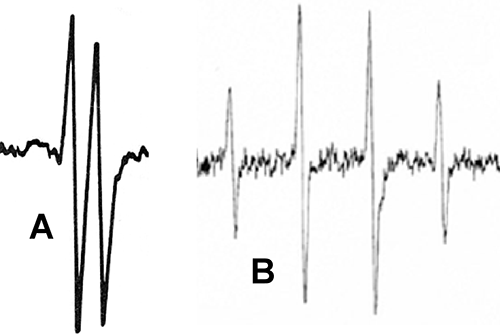
Fig. III-13 A : Spectre RPE du radical ascorbyle ; formule : voir fig. IV-9
B : Spectre RPE d'un adduit de spin.
3. Limites de la RPE : la sensibilité moyenne (1014 centres détectables dans la cavité) est relativement faible, mais spécifique.
Il est probable que la sensibilité pourra être améliorée avec les nouvelles techniques de génération de microondes. Mais la préparation relativement longue des échantillons avant l'examen en RPE empêche de saisir l'apparition de radicaux fugaces à température ordinaire. Dès les débuts de la RPE, les chercheurs eurent recours au "gel" de ces radicaux en plongeant les échantillons dans l'azote liquide (- 273°C) et en thermostatant la cavité de résonance à cette température par une circulation de l'azote liquide. On recourt même à présent à l'hélium liquide.
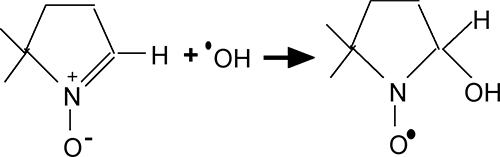
Fig. III-14 : Structure et réaction d'un "spin-trap" classique : le DMPO.
Le stratagème des adduits de spin: dès son apparition, le radical est mis en contact avec une nitrone. La nitrone est une molécule azotée qui, au contact d'un radical, réagit avec lui en captant son électron célibataire et en devenant un radical très stable (fig. III-14), grâce à une transformation de structure. Ce radical témoigne de l'existence brève d'un radical naturel (s'il s'agit d'un échantillon de tissu vivant), mais sa spécificité laisse à désirer.
6. Les oxydo-réductions: Généralités
1) Les différences dans l'attirance des électrons
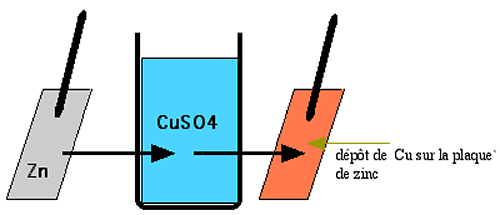
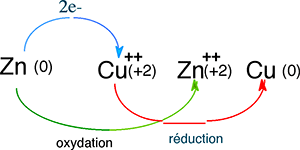
Fig. III-15 : Captation des électrons du Zn par le cuivre Cu++.
Si l'on plonge une plaque de zinc dans une solution de sulfate de cuivre (de préférence concentrée, pour la rapidité de constatation du phénomène), cette plaque perd sa couleur grise et devient rougeâtre par dépôt de cuivre métallique :
Zn + Cu++ ==> Zn++ + Cu
Le zinc est plus réducteur que le cuivre auquel il a donné 2 électrons. Le cuivre attire plus les électrons que le zinc : il est plus oxydant.
Au cours de l'échange d'électrons entre deux atomes ou molécules, la molécule donneuse d'électron(s) s'oxyde, tandis que l'autre se réduit. On dit aussi que le nombre d'oxydation de la première s'élève, en même temps que celui de la deuxième s'abaisse. Dans le phénomène ci-dessus, le cuivre montre son caractère plus oxydant que le zinc.2) Les piles
Les différences d'affinité pour les électrons sont à l'origine du courant électrique des piles (Fig. III-16A). La pile de Daniels est une application de l'expérience de la fig. III-15.
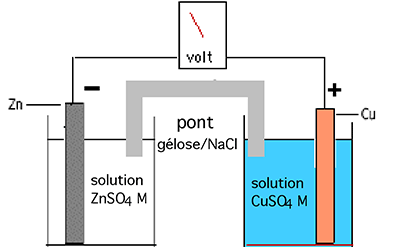
A B
Fig. III-16 : A. Pile expérimentale de Daniels. On dispose de 2 vases contenant chacun :
1) Une solution de sel du premier métal dans lequel est plongée une tige de ce métal. Dans cet exemple, il s'agit d'une électrode de zinc immergée dans une solution de sulfate de zinc.
2) Une solution de sel de l'autre métal, à la même concentration molaire qu'en 1) et une électrode de ce métal.
Le sel utilisé ici est le sulfate de cuivre et l'électrode est en cuivre.
Pour qu'un courant électrique (déplacement d'électrons) se produise, il faut constituer un circuit qui sera réalisé:
1) par un conducteur métallique traversant un voltmètre et réunissant les extrémités non immergées des deux électrodes;
2) par un pont semi-liquide conducteur (gelée salée) réunissant les deux solutions.
Le voltmètre aura une très haute résistance d'entrée afin que l'intensité du courant électrique généré soit très faible et que la différence de potentiel mesurée soit proche de la valeur théorique.
B. Schéma des échanges d'électrons : Ces réactions sont étroitement couplées et ne peuvent exister l'une sans l'autre. Les électrons d'échange restent étroitement liés à leur noyau. On ne peut les imaginer libres.
3) Spontanéité d'une oxydo-réduction : Equation de Gibbs
Les réactions spontanées sont celles qui dégagent de la chaleur (réactions exothermiques) tandis que celles qui en absorbent (endothermiques) dépendent d'un apport énergétique externe. La thermodynamique nous apprend que ces phénomènes sont associés à une variation d'énergie libre (ΔG) dans le système réactionnel, et qu G est négative dans les réactions spontanées et positive dans les réactions endothermiques.
ΔG est liée à la variation du potentiel d'électrode (ΔE) par la relation (découlant de l'équation de Gibbs) :ΔG= - nF ΔE
(n = le nombre d'électrons mis en jeu et F la charge en faradays, ΔE étant la variation de potentiel d'électrode exprimée en volts).
La démonstration de cette équation dépasse largement le cadre de cette introduction, mais nous amène à admettre que la spontanéité d'une réaction est liée au caractère négatif de ΔG, donc au caractère positif de la variation du potentiel d'électrode ΔE.
Nous retiendrons l'exemple de la réduction de l'oxygène en anion superoxyde. Pour passer de O2 à O2•, à 20°C et à la pression atmosphérique, il y a une variation de potentiel de - 0,33 V (comme nous l'apprend l'expérience). Le calcul du ΔG donnera donc une valeur positive, ce qui signifie que
O2 + e- => O2• est une réaction endothermique (non spontanée)et que pour se produire, elle doit être catalysée (par voie enzymatique, par exemple).
Ceci est une autre démonstration de l'inertie chimique de l'oxygène dans les conditions biologiques.
4) Potentiel normal
Les variations d'affinité des éléments et substances chimiques se mesurent par les potentiels d'électrode.
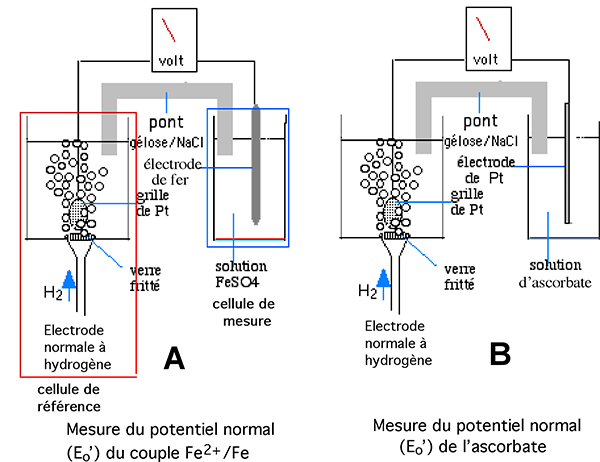
Pour constituer une échelle de comparaison des divers potentiels, on a imaginé de les comparer à une électrode standard, servant de référence, l'électrode standard à l'hydrogène (Electrode Normale d'Hydrogène : ENH), dont le potentiel constituera le point zéro dans une gamme de potentiels positifs et négatifs. Un schéma de l'ENH est représenté sur la fig. III-17. Dans une cellule électrolytique contenant une solution d'électrolyte, on réalise un dégagement continu d'hydrogène sous une grille d'un métal pratiquement inerte électrochimiquement, le platine. De l'hydrogène est ainsi adsorbé sur le platine et constitue la partie active de l'électrode normale.
Pratiquement, on constitue une pile selon le modèle de la figure III-17 A où la cellule de référence est constituée par l'électrode normale à hydrogène. Celle-ci est reliée à la cellule de mesure par un montage potentiométrique et par un pont conducteur.
Fig. III-17 A : L'électrode normale à hydrogène constitue la cellule électrochimique de référence.
Pour mesurer le potentiel du couple Fe/Fe2+, on constitue une cellule de mesure où une tige de fer plonge dans une solution de sel de fer ferreux. Ce montage est valable pour les autres métaux et leurs sels.
Fig. III-17 B : Montage pour la détermination de Eo' d'un composé soluble, tel l'ascorbate. L'électrode de mesure est ici un fil de platine.
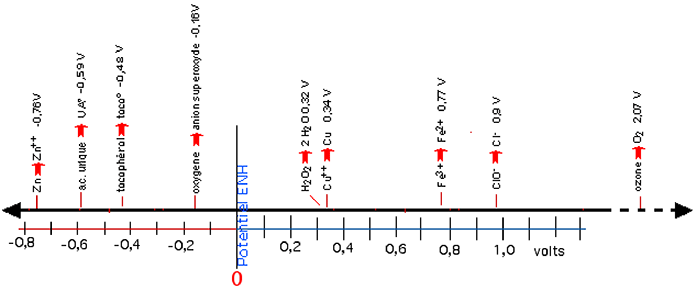
5) Echelle d'oxydo-réduction
On a établi sur ce concept d'électrode au potentiel zéro l'échelle horizontale de la fig. III-18 où se trouvent à gauche (rouge), les corps les plus réducteurs, et à droite (bleu), les plus oxydants.
Fig. III-18 : Exemple d'échelle d'oxydo-réduction établie en fonction de l'électrode à hydrogène. Les valeurs sont exprimées en volts.Dans la pile de Daniels (fig. III-16), la différence de potentiel mesurée par le voltmètre est de 1,1 volts. Cette valeur se retrouve en additionnant les valeurs absolues (figurant sur l'échelle ci-dessus), calculées pour le zinc et le cuivre :
0,76 V + 0,34 V = 1, 1 V
6) Les nombres d'oxydation
Une équation redox simple, mais très importante en biochimie de l'oxygène, est la suivante (fig. III-19), qui met en évidence la conservation du nombre total d'oxydation dans chaque membre des équations redox : c'est la réduction du fer ferrique Fe3+ en fer ferreux Fe2+ par l'anion superoxyde :
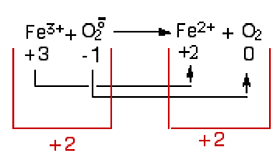
Fig. III-19 : Réduction du fer ferrique par l'anion superoxydeDans cette réaction, le nombre d'oxydation du fer est passé de +3 à +2, tandis que celui de l'oxygène de -1 à 0. Le nombre d'oxydation total est conservé.
Il y a oxydation d'un élément lorsque son nombre d'oxydation croît (ici : l'anion superoxyde) et réduction lorsque son nombre d'oxydation diminue (cas de Fe3+).7) Cas des molécules
Nous verrons plus loin dans ce chapitre que des électrons s'échangent entre molécules organiques. Un donneur d'électron D peut transférer un ou des électrons à un accepteur A, sans qu'il y ait changement du nombre d'oxydation des atomes constitutifs (ex: le glutathion GSH).
D est le réducteur et A l'oxydant. Autrement dit, D est oxydé tandis que A est réduit.
Oxydation et réduction sont simultanées et indissociables8) Remarque sur l'aliénation des électrons
Les électrons libres n'existent pas en dehors du plasma (4ième état de la matière, à hautes températures).
Les électrons sont confinés dans un puits de potentiel d'une profondeur de 13,5 électron-volts.
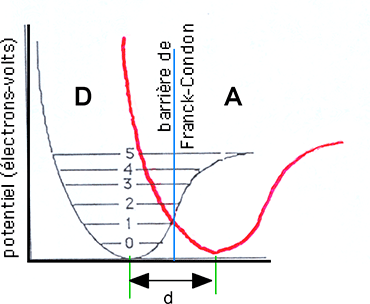
Fig. III-20 : En gris : le puits de potentiel d'un donneur d'électron D (les différents niveaux d'énergie sont figurés). En rouge, le puits de potentiel d'un accepteur d'électron A. d (en angströms) représente la distance internucléaire. Pour qu'un électron puisse passer de D à A, il doit franchir la barrière de Franck-Condon, grâce à une énergie provenant par exemple d'une différence de potentiel rédox ou d'un rayonnement.
7. Bibliographie
Principe de Pauli, multiplicité, résonance paramagnétique électronique :
Leffler JE. An introduction to free radicals, John Wiley & Sons, New York, 1993, p.34-76.Réactions radicalaires : Bauld NL. Radicals, ion radicals and triplets. Wiley-VCH, New York, 1997, pp. 41-76.
Oxydo-réductions: Généralités
Glasstone S. Textbook of Physical Chemistry. Mac Millan édit, ré-édition de 1969, chap. X (electrolysis), pp. 884-953.Electrochimie de l'oxygène
Sawyer DT. Oxygen Chemistry. Oxford, University Press, 1991, pp.19-50.